ALK(AnaplasticLymphomaKinase,間變性淋巴瘤激酶)是一種受體酪氨酸激酶,屬于胰島素受體超家族。ALK高度保守,主要在成人腦組織中表達,對神經系統的發育起重要作用。1994年,科學家們首次在間變性大細胞非霍奇金淋巴瘤(ALCL)中發現了由染色體易位形成的(NPM)-ALK融合蛋白,隨后,ALK與其他基因的融合被證實存在于ALCL、炎性肌纖維母細胞瘤、彌漫性大B細胞淋巴瘤、鱗狀細胞癌和非小細胞肺癌中。除染色體易位之外,ALK基因擴增和野生型ALK蛋白的點突變激活也被報道存在于成神經細胞瘤、卵巢癌和炎性乳腺癌中,這使得ALK成為了各種含ALK融合的血液疾病和實體腫瘤治療的一個有吸引力的靶點。
目前,已經上市和正處于臨床試驗階段的ALK抑制劑有:克唑替尼(1,crizotinib)、色瑞替尼(2,ceritinib)、阿來替尼(3,alectinib)、布加替尼(4,brigatinib)、勞拉替尼(5,lorlatinib)、ASP3026(6)(圖1)。
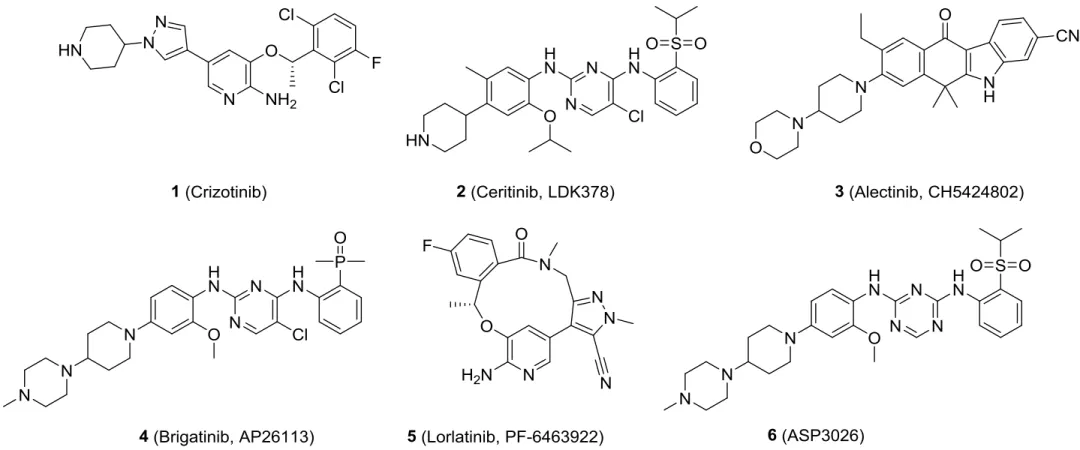
圖1 已經上市和處于臨床階段的ALK抑制劑
其中,crizotinib是FDA批準的第一款ALK抑制劑,用于ALK陽性非小細胞肺癌(NSCLC)患者的臨床治療,但大部分患者在1~2年用藥后容易產生耐藥突變或轉移導致病情惡化。隨后,ceritinib、alectinib、brigatinib和lorlatinib先后獲批用于晚期轉移性的NSCLC治療。ASP3026目前正處于臨床試驗中。
近期,王少萌團隊發現了一種新的高效、口服活性的ALK抑制劑CJ-2360,它能有效抑制野生型ALK激酶和幾種臨床報道的ALK突變體,并能在ALK-陽性的KARPAS-299移植瘤模型中實現完全的腫瘤消退(圖2)。
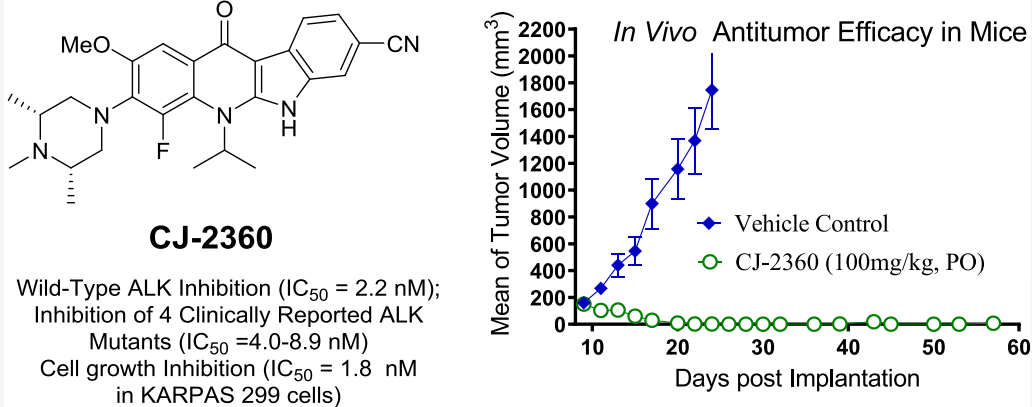
圖2 CJ-2360的活性評估數據
在所有已獲批上市或正處于臨床階段的ALK抑制劑中,alectinib是唯一一個具有四環并環結構的分子,并且其已被證明對ALK的選擇性優于其他ALK抑制劑。因此,研究者選擇alectinib作為設計新ALK抑制劑的模板。
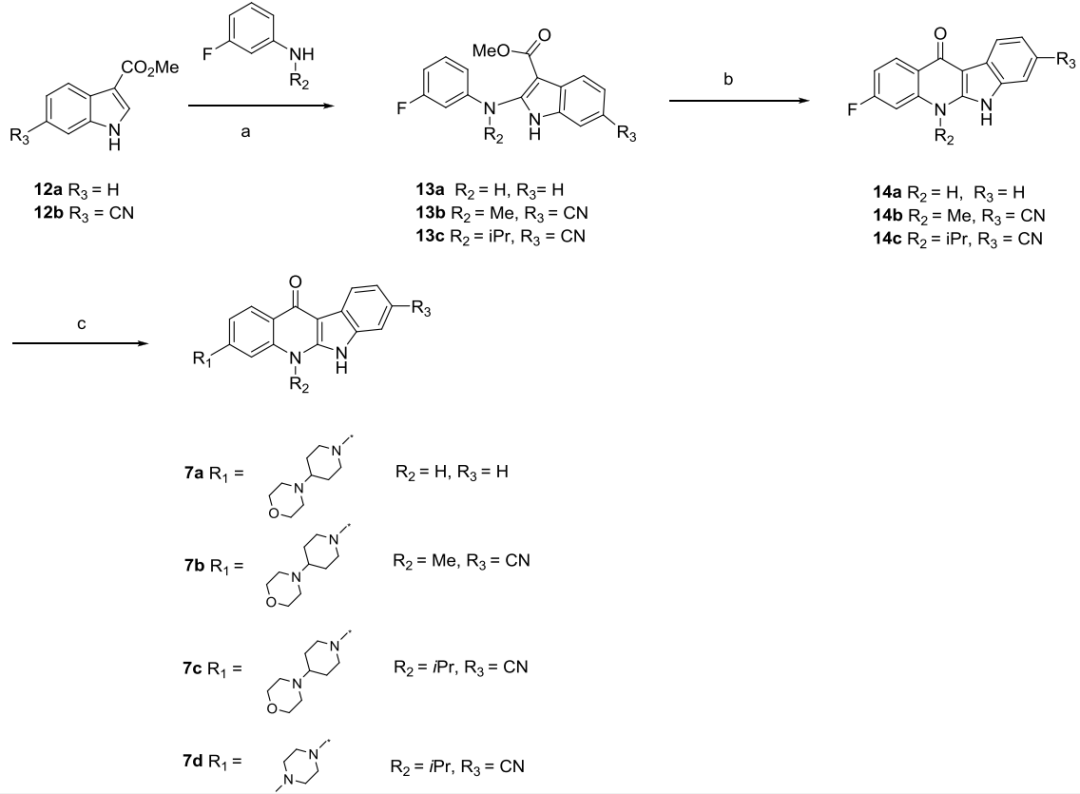
為了保留四環并環的結構,研究者以吲唑并喹啉為主體骨架設計了化合物7a和7b(圖3)。在體外酶活性實驗中,7a即使濃度達到50μM也沒有表現出抑制活性,而7b的IC50值為2.5μM。在alectinib結構中,羰基對面有兩個甲基,研究者將7b相應位置的甲基替換成異丙基得到化合物7c,7c的IC50值達到28.6nM。進一步對7c的主要代謝位點進行改造,將4-嗎啉基哌啶基替換成1-甲基哌嗪基得到了活性更好的化合物7d(圖3)。
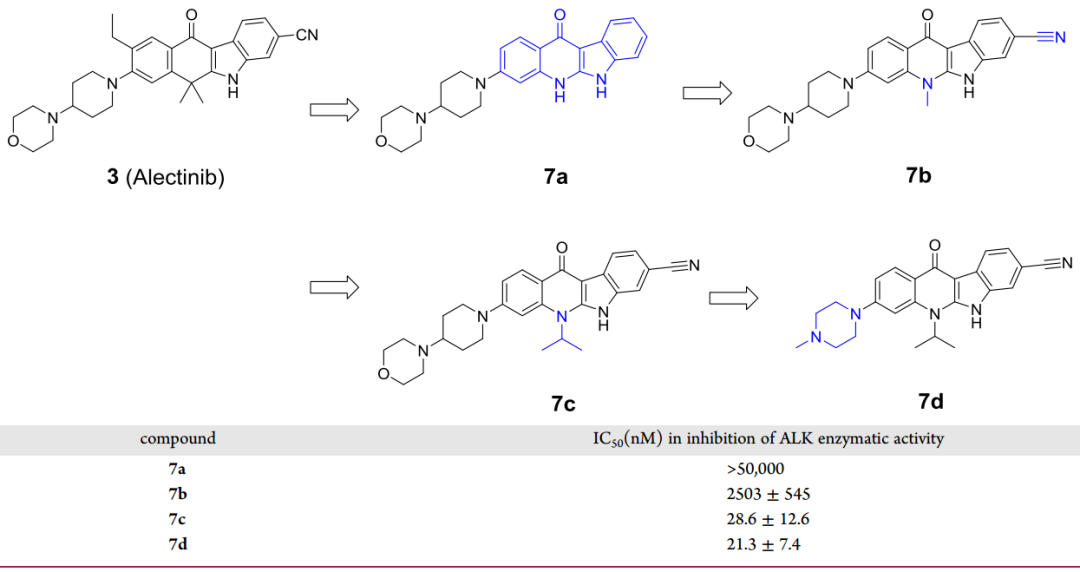
圖3 化合物7a-7c的設計及活性數據
參照alectinib與ALK形成的復合物的共晶結構,研究者建立了化合物7d與ALK的結合模型(圖4),兩者有相似的結合模式,比較大的區別在于,alectinib的四環并環骨架上有一個乙基與ALK上的L1198、A1200和L1122殘基存在疏水相互作用,而化合物7d在相同的位置上沒有任何取代基。
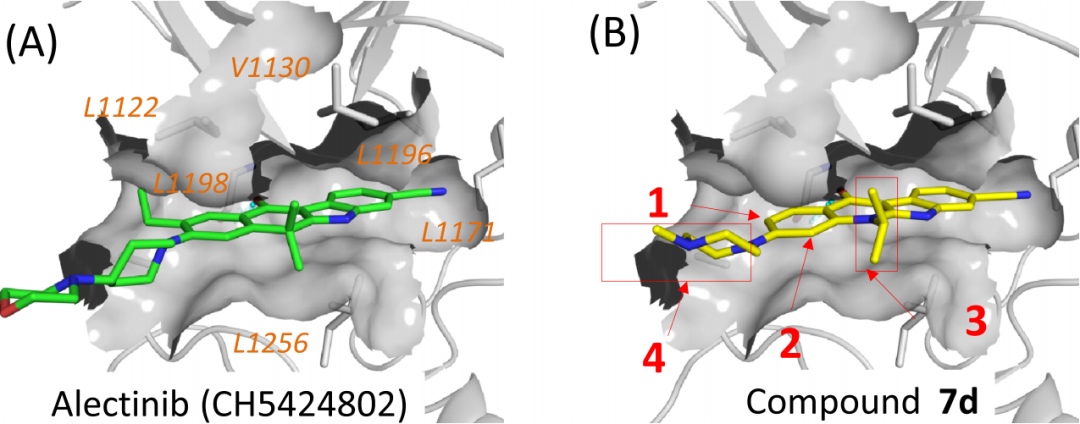
圖4 ALK分別與alectinib和化合物7d形成的復合物結構
如圖4-B箭頭所示,在化合物7d的1號位置上引入一個乙基得到化合物8b,其抑制活性相比7d有了10倍的提升(圖5-8bvs7d),除了異丙基取代外,其它的烷基取代或溴取代均給出了不錯的IC50值(圖5-8a-8e)。研究者繼而將烷基取代替換成烷氧基取代合成了化合物8f-8i,這些化合物對ALK的抑制能力都要優于7d。更重要的是,研究者在攜帶ALK融合蛋白的KARPAS-299細胞系中評估了上述化合物對細胞增殖的抑制活性,其效果相比7d均有較大提高。
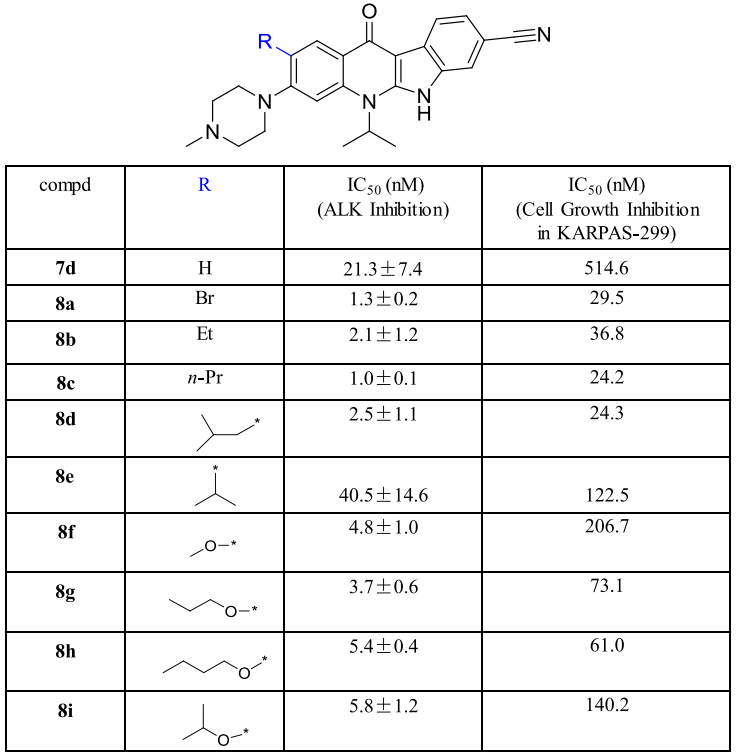
圖5 對化合物7d的1號位改造
如圖4-B箭頭所示,在化合物7d的2號位置上引入氟原子得到化合物9a,其對ALK的抑制能力與7d相差不大,但在KARPAS-299細胞系中,9a的活性比7d提高了10倍(圖6)。以8f為模版化合物,在其2號位引入氟原子得到化合物9c,其細胞活性相比8f也有10倍的提升。這些數據表明,在苯環上引入氟原子可以顯著提高化合物的細胞效力。
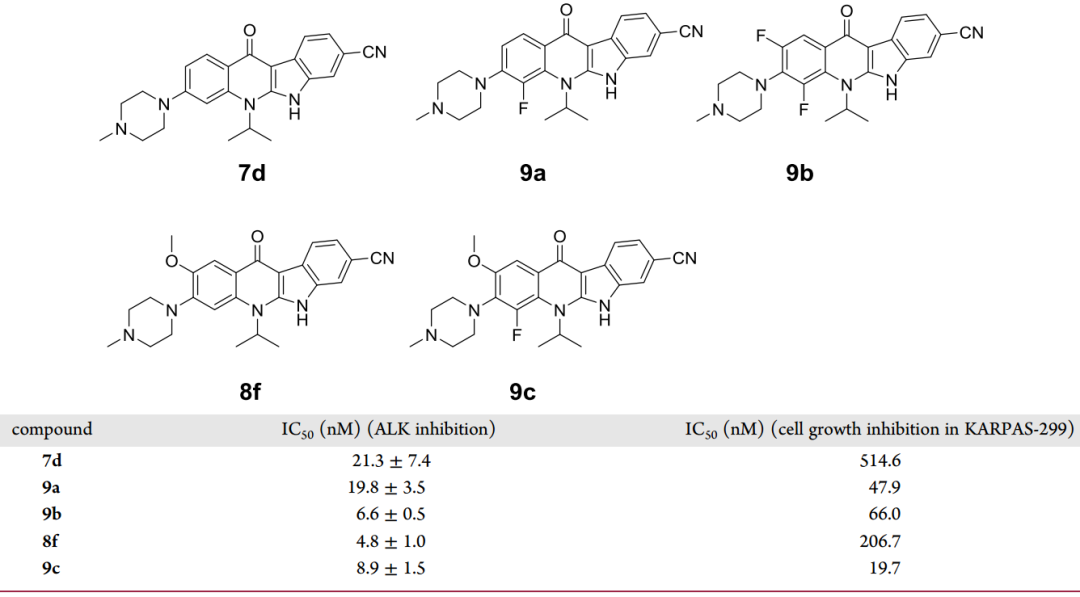
圖6 對化合物7d的2號位改造
以化合物8f為模版,對喹啉氮上的取代基進行考察(圖7),將異丙基替換成其它脂肪鏈如乙基、丙基、仲丁基或2-戊基得到化合物10a-10d,這些化合物對ALK的抑制活性相比8f均有不同程度的下降;將異丙基替換成環烷基,特別是環丁基、環戊基或環己基時,它們在KARPAS-299細胞系中表現出非常相似的細胞增殖抑制活性。
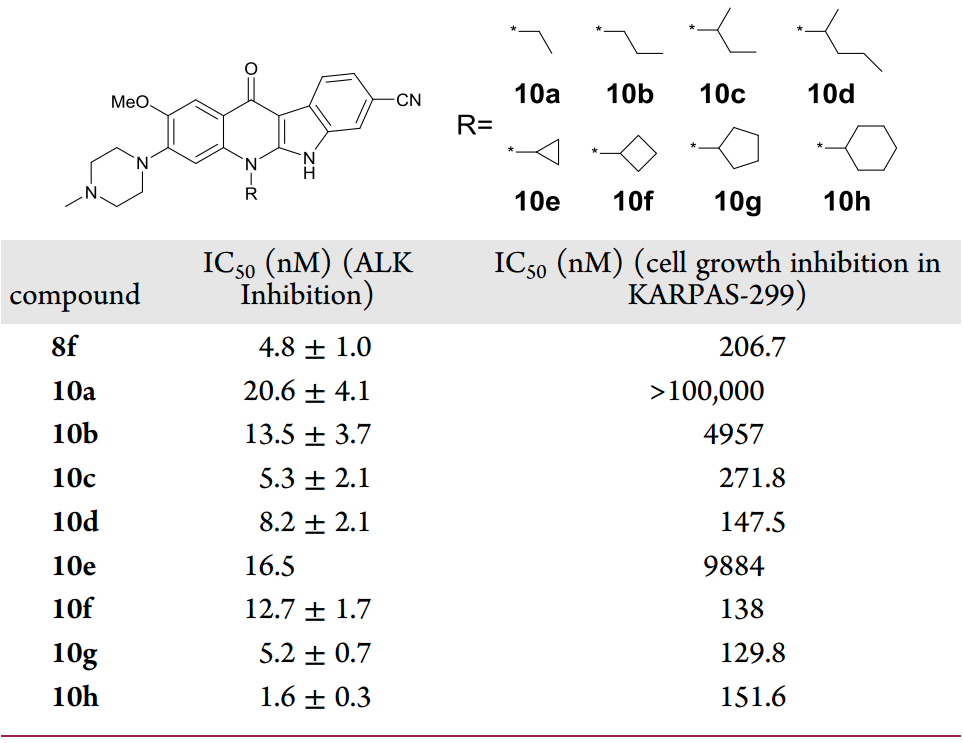
圖7 對化合物8f的3號位改造
最后,研究者以9c為模版,考察了4號位不同增溶基對分子活性的影響(圖8),最終得到活性最好的化合物CJ-2360,其不僅表現出很強的ALK抑制能力,在KARPAS-299細胞系中抑制細胞增殖的IC50值也達到了1.8nM。
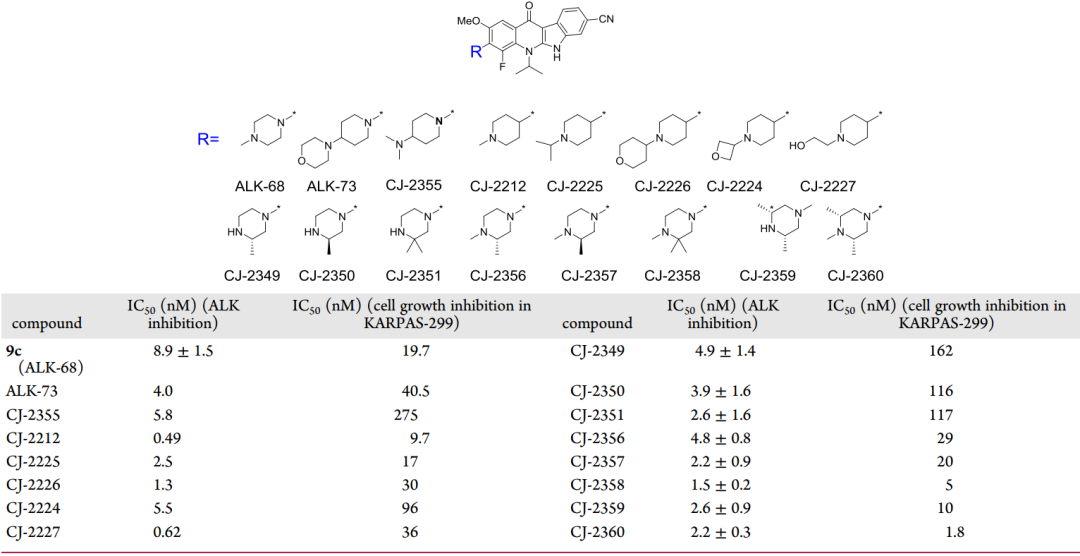
圖8 對化合物9c的4號位改造
在眾多考察的化合物中,CJ-2360表現出了綜合最好的酶活性和細胞活性,隨后,研究者評估了CJ-2360分別在小鼠和大鼠體內的藥代動力學參數(圖9)。結果顯示,在口服給藥后,CJ-2360在小鼠體內達到了合理的口服暴露量,整體的口服生物利用度為38.2%,中等的血漿清除率(1.2L/h/kg),較大的容積分布(7.5L/kg)表明CJ-2360在小鼠組織分布廣泛。在大鼠體內的藥代動力學研究給出了相似的數據。
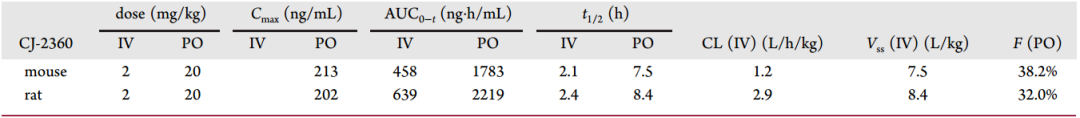
圖9 CJ-2360在小鼠和大鼠體內的藥代動力學參數
此外,研究者對攜帶KARPAS-299移植瘤的小鼠進行口服給藥,并分析了CJ-2360在小鼠血漿和腫瘤組織中的藥物濃度,證實CJ-2360在小鼠單次口服給藥后便獲得良好的血漿暴露量,而且其在腫瘤組織中的濃度要遠大于在血漿中的濃度。
基于CJ-2360良好的體內藥代動力學數據和腫瘤組織暴露量,研究者評估了其在攜帶KARPAS-299移植瘤小鼠體內的抗腫瘤效果,并以ceritinib作為陽性對照(圖10)。數據顯示,CJ-2360在每天兩次給藥后能夠實現所有腫瘤完全消退,直到最后一次給藥后的第23天腫瘤才開始恢復,證明CJ-2360在攜帶KARPAS-299移植瘤小鼠體內能夠實現完全且持久的腫瘤消退,此外,小鼠對CJ-2360耐受性良好,在整個實驗過程中沒有引起體重減輕或其他毒性癥狀。作為對比,ceritinib只能實現部分腫瘤的完全消退。
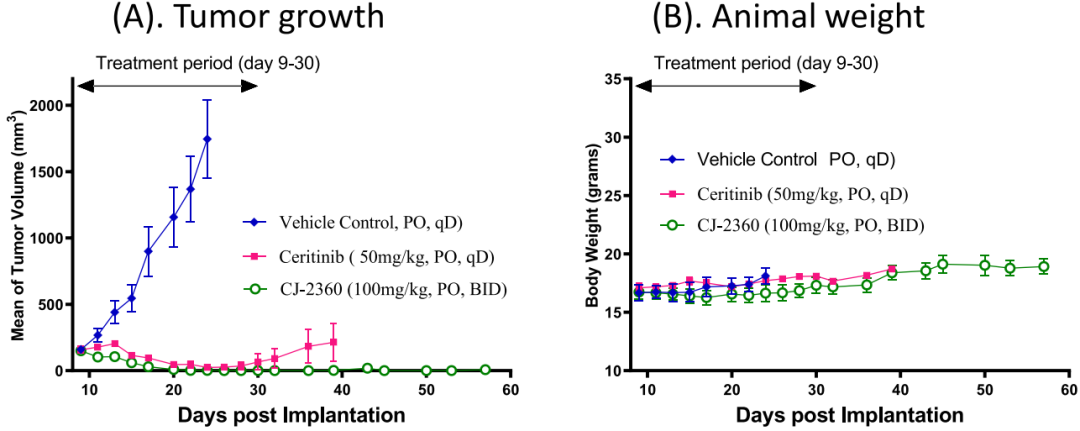
圖10 CJ-2360的體內抗腫瘤活性評估
臨床中,部分ALK陽性腫瘤患者在經crizotinib治療后,由于ALK突變而產生耐藥性,目前已鑒定出幾種臨床ALK突變體。在ALK抑制劑的研發過程中,其對各種ALK突變體的抑制能力是必須考察的,因此,研究者對比了幾種已經上市的ALK抑制劑和CJ-2360對ALK突變體的抑制活性(圖11)。結果表明,CJ-2360對包括F1197M、G1269A、L1196M和S1206Y在內的ALK突變體也具有很高的抑制活性,分別達到了4.0、8.8、6.3和8.9nM。
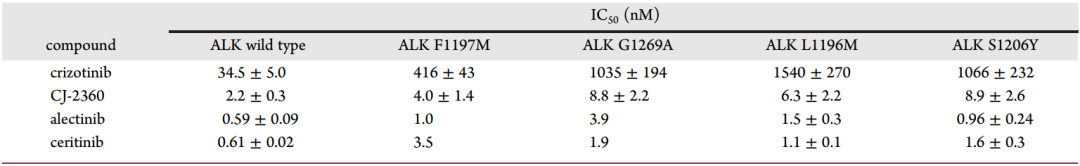
圖11 幾種抑制劑對ALK野生型和突變體的抑制能力對比
研究者以alectinib為模板化合物,設計合成了一系列包含吲唑并喹啉骨架的新型ALK抑制劑,通過評估最終篩選出綜合酶活性和細胞活性最好的化合物CJ-2360。CJ-2360有著良好的口服血漿暴露量、腫瘤組織濃度以及生物利用度,對野生型ALK和幾種臨床ALK突變體均有很高的抑制活性。最重要的是,體內抗腫瘤實驗證實CJ-2360能實現包括野生型和突變型在內的所有ALK陽性腫瘤的完全持久消退,這一結果要優于ceritinib,這些實驗表明CJ-2360是一種有望用于晚期臨床前研究的新型ALK抑制劑。
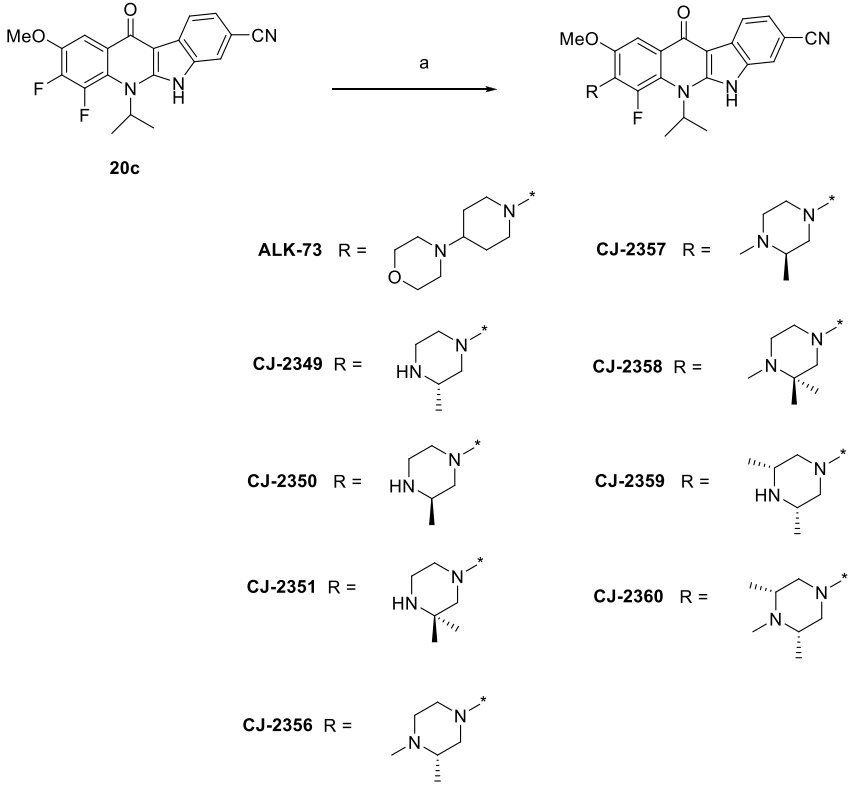
6、
合成ALK
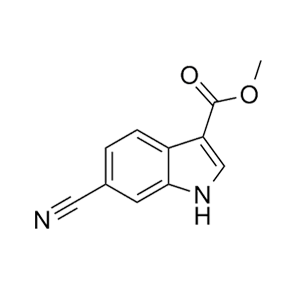
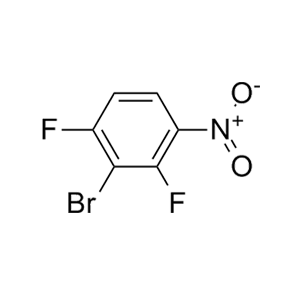
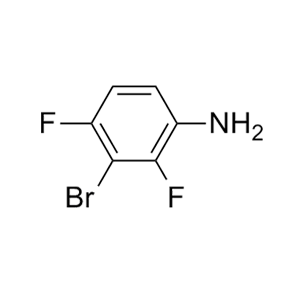
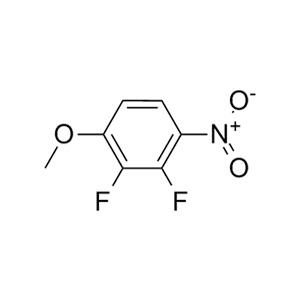
-73、CJ-2349-2351和CJ-2356-2360
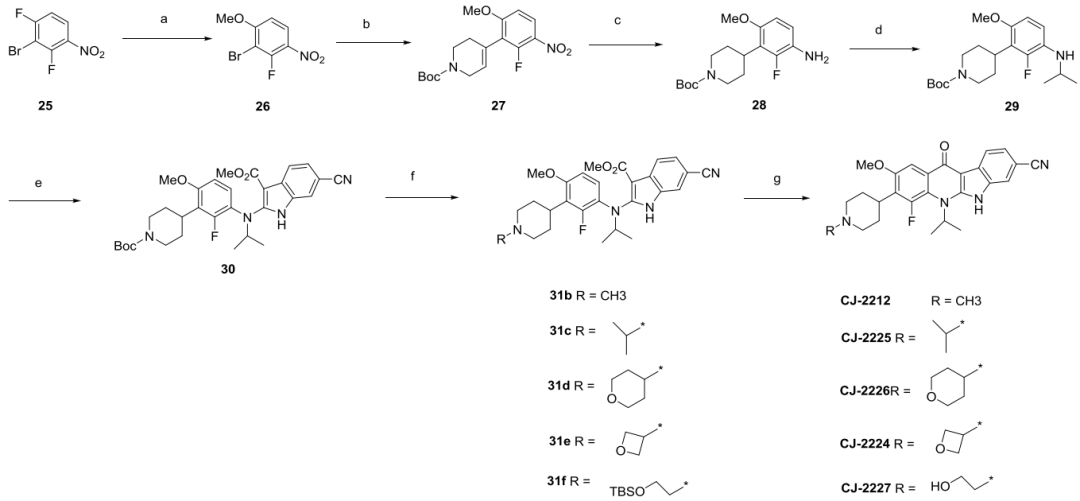
7、
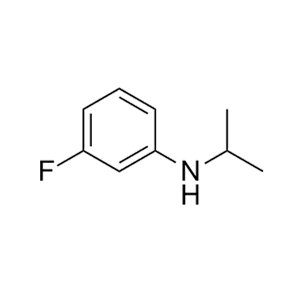
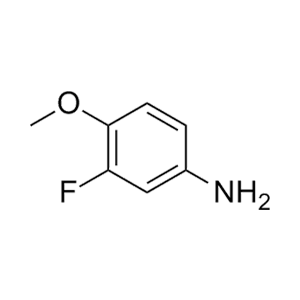
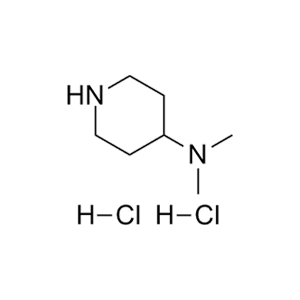
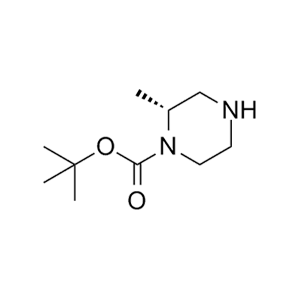
合成CJ-2212和CJ-2224-2227
JianyongChen, Yunlong Zhou, Xuyuan Dong, Liu Liu, Longchuan Bai, DonnaMcEachern, Sally Przybranowski, Chao-Yie Yang, Jeanne Stuckey,Xiaoqin Li, Bo Wen, Ting Zhao, Siwei Sun, Duxin Sun, Lingling Jiao,Yu Jing, Ming Guo, Dajun Yang, and Shaomeng Wang Discovery of CJ-2360as a Potent and Orally Active Inhibitor of Anaplastic Lymphoma KinaseCapable of Achieving Complete Tumor Regression Journalof Medicinal Chemistry(2020),63(22),13994-14016.
注:文中圖片均來源于參考文獻
 技術支持
技術支持



 400-821-0725
400-821-0725

 滬ICP備17019645號
滬ICP備17019645號







