囊性纖維化(cysticfibrosis,CF)是一種危及生命的隱形遺傳性疾病,由囊性纖維化跨膜電導調節因子(cysticfibrosis transmembrane conductanceregulator
,
CFTR)
基因突變導致CFTR蛋白功能缺陷或缺失所致。
CFTR的突變影響各種組織,包括肺、胰腺、腸道、生殖道和汗管,使囊性纖維化成為一種全身性疾病。
其最嚴重的影響是導致呼吸道中氯化物的分泌減少,隨后的肺表面脫水導致肺內黏液厚度增加,致使氣道粘液纖毛清除受損,進而導致氣道阻塞、慢性細菌感染和過度炎癥。
最終,肺損傷導致肺功能進行性下降。
至今為止,已知的CFTR突變超過2000種,最常見的CFTR突變是F508del突變(多肽鏈位置508處的苯丙氨酸缺失),該突變使CFTR的折疊功能受損,最終導致細胞表面存在的離子通道數量減少。
近年來,一系列的調節劑被應用于攜帶CFTR突變患者的治療中,這些CFTR調節劑包括校正劑和增效劑(通過增加細胞表面CFTR蛋白的門控活性(離子跨膜的能力)來增加缺陷型CFTR蛋白的功能),前者可以提高細胞表面CFTR水平,后者可以改善CFTR離子通道的門控功能。目前,已經批準用于CF患者治療的CFTR調節劑有:增效劑Ivacaftor;校正劑Lumacaftor、Tezacaftor和Elexacaftor(圖1)。對于攜帶F508delCFTR純合子突變的患者,已經有校正劑與Ivacaftor增效劑聯合療法,而攜帶G551D等門控突變的患者細胞膜上已經有足夠的CFTR,因此可以使用Ivacaftor作為單藥治療。
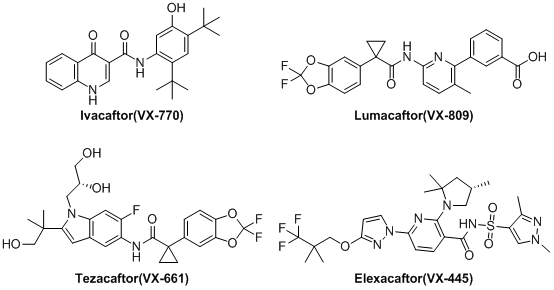
圖1 已批準用于CF治療的校正劑和增效劑
2018年,比利時生物制藥公司GalapagosNV的科學家們報道了一種增效劑GLPG1837(圖2),雖然GLPG1837與Ivacaftor在結構上有很大的不同,但它們競爭的結合位點是相同的,而且這兩種化合物都需要每天兩次給藥,以提供足夠的血漿暴露量。近日,該公司團隊通過進一步篩選優化發現了基于藥代動力學結果的僅需每日一次給藥的新型增效劑GLPG2451。
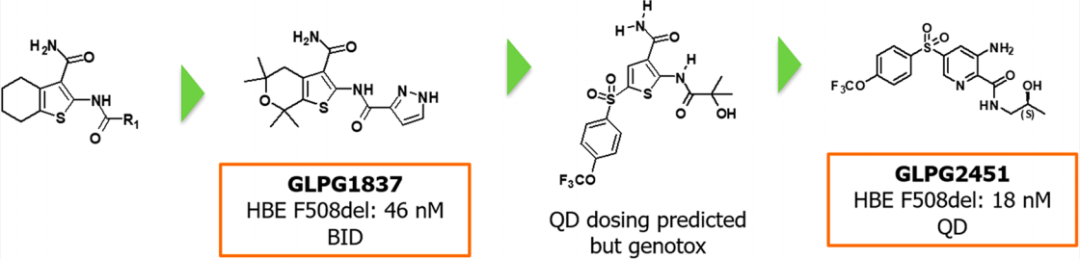
圖2 新型增效劑GLPG2451的發現過程
增效劑GLPG1837的發現是基于已存在的增效劑的結構,對589種商業化分子進行篩選的結果,此外,科學家們還發現GLPG1837上的環己基也可以被磺酰胺基團取代,繼而發現了化合物4,并測得其對F508del突變體的EC50為17nM(圖3)。在攜帶G551D/F508del突變患者的人原代支氣管上皮細胞(HBE)實驗中證實,化合物4和GLPG1837對G551DCFTR通道的增強程度均明顯高于Ivacaftor。
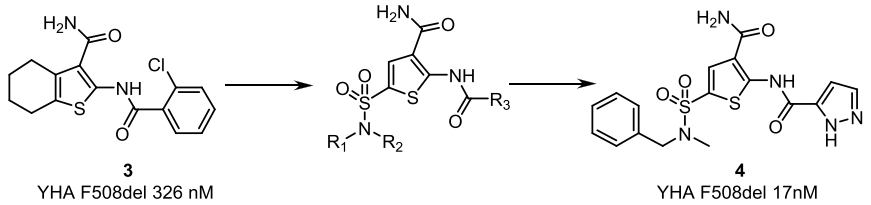
圖3 化合物4的發現過程
通過大鼠微粒體分析,針對清除率對化合物4的結構進行優化,將與磺酰基連接的胺替換成哌啶得到化合物5,其清除率明顯下降。提高化合物親脂性,將磺酰胺部分替換成砜得到化合物6,由于親脂性的增加通常導致清除率的增加,而化合物6的實驗結果表明,砜作為連接子有其固有的穩定性(圖4)。
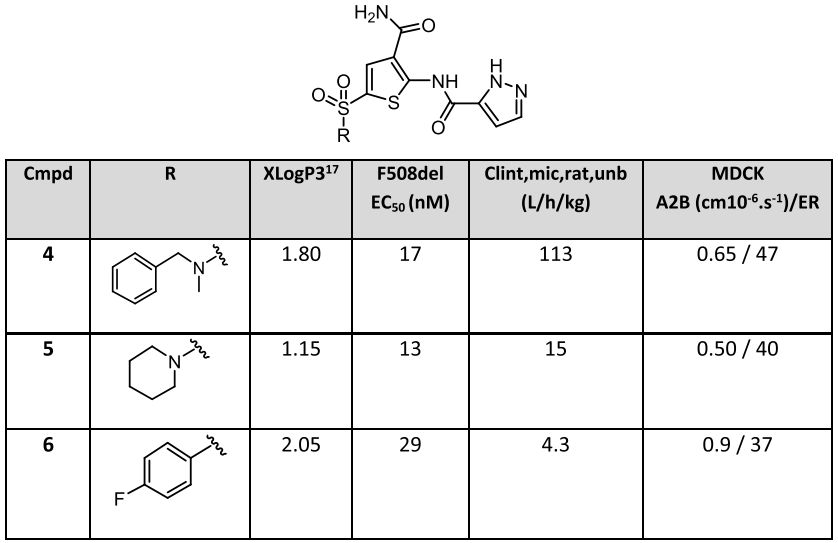
圖4 磺酰胺部分優化
從圖4中可以看到,當磺酰胺或砜結構與吡唑酰胺同時存在時,分子具有較低的被動滲透性和高通量比,將吡唑基替換成叔丁基得到化合物7(圖5),化合物7的細胞活性大大下降,可能是因為其上缺少氫鍵供體和受體,在此基礎上設計合成了化合物8和9,這兩種化合物的通量比差異較大,經過構象分析,化合物9相比化合物8,沒有環丙基約束,使得其羥基與羰基形成氫鍵后能夠有效屏蔽極性中心。而且化合物9在大鼠體內的血漿暴露量遠遠高于化合物8。
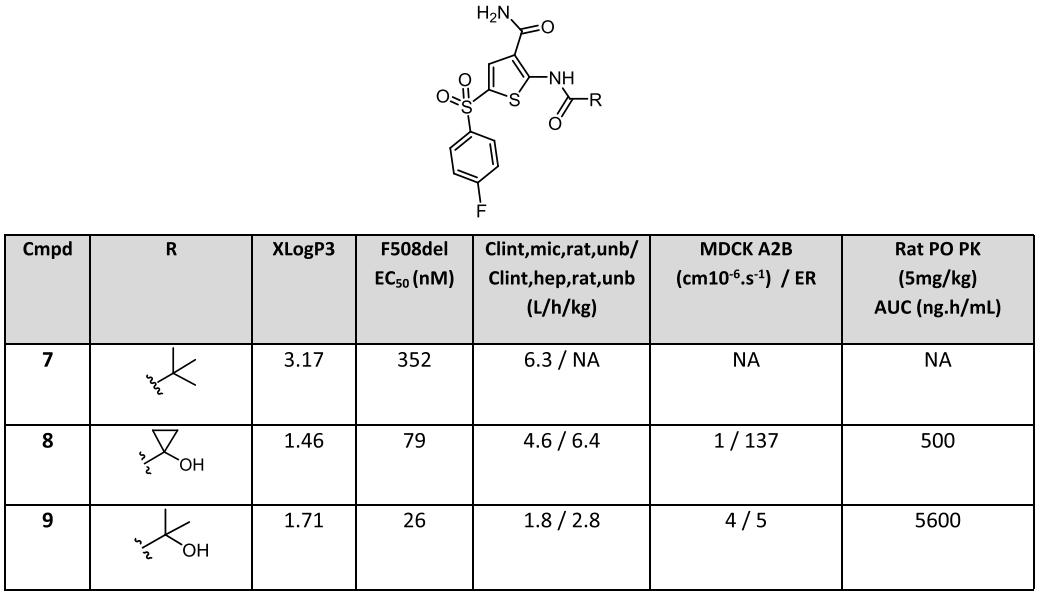
圖5 酰胺部分優化
盡管對F508del突變體的活性是研究一系列化合物SAR的主要參考因素,但在分子優化后期,基于G551DCFTR上的活性數據是進一步區分它們的重要依據。研究發現,通過改變砜的苯環上的取代基能進一步提高分子的效能,由此得到化合物10,雖然分子整體的親脂性提高,但化合物10在大鼠和人肝細胞中依然表現出較低的清除率,而且在大鼠體內展示出令人滿意的藥代動力學數據(圖6)。
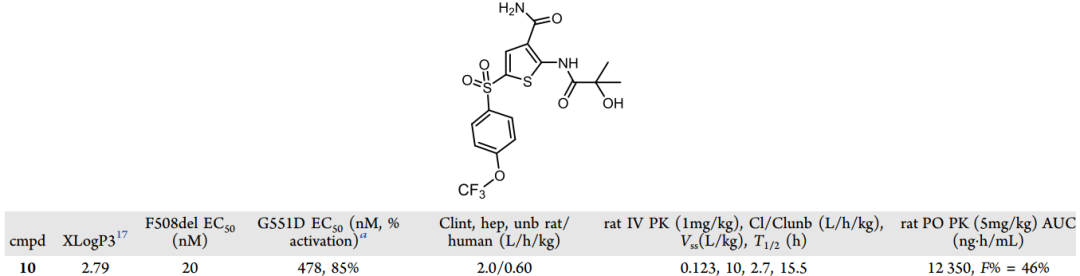
圖6 化合物10的實驗數據
相比化合物9,化合物10不僅提高了對G551DCFTR的效力,也將原來需要每天兩次給藥的方案改善至每天一次給藥。雖然化合物10表現出令人滿意的臨床前結果,但進一步的研究發現包括化合物10在內的多種類似物的體外微核試驗呈陽性,因而需要繼續篩選出沒有基因毒性的新化合物。
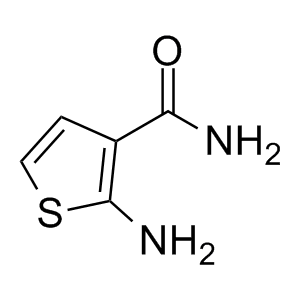
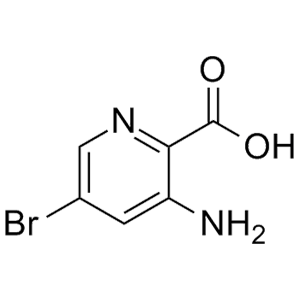
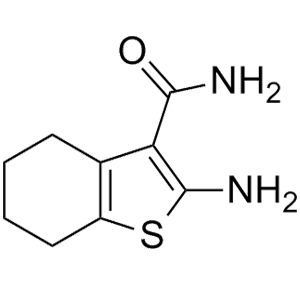
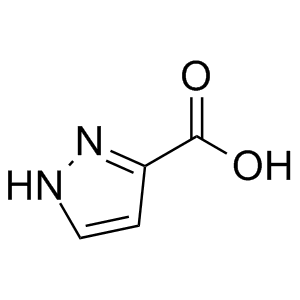
在化合物篩選過程中,無論是噻吩左邊SO2連接部分還是右邊酰胺部分都已經進行了廣泛的研究,兩者均被認為是值得保留的結構。化合物10的分子內相互作用模型表明(圖7),分子內存在廣泛的接觸,中央部分可以認為基本是平面結構,基于這樣的理解,將噻吩環換成吡啶環、甲酰胺換成氨基并將與雜芳環連接的酰胺鍵反轉設計得到化合物11,兩種化合物可以被視為一對匹配分子,它們保持相似的分子內相互作用,并且具有相似的電荷分布(圖7-A)。
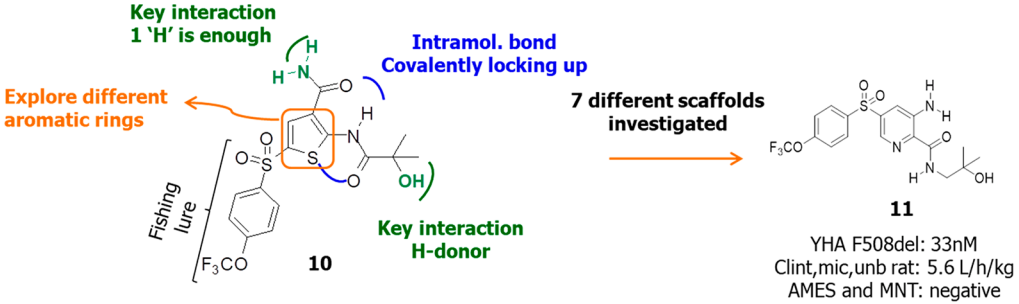
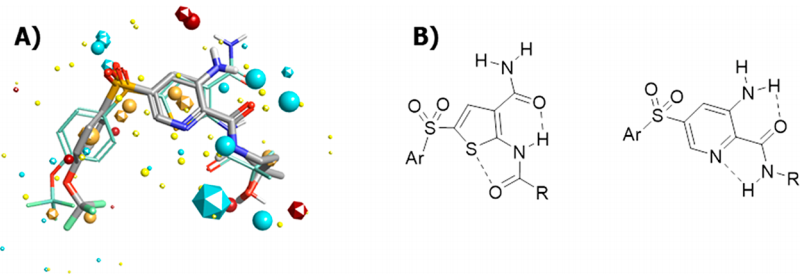
圖7 基于化合物10的主要骨架設計化合物11
數據顯示,化合物11為對F508delCFTR的EC50為33nM,更重要的是,化合物11的體外微核試驗(MNT)和污染物致突變性試驗(AMES)均呈陰性,證明無基因毒性。在此基礎上,考察了化合物11中與SO2連接的芳環部分和與CO連接的胺部分(圖8)。值得注意的是,當酰胺上的N被完全烷基化時,化合物的活性喪失,這與分子設計模型是一致的(圖7-B)。
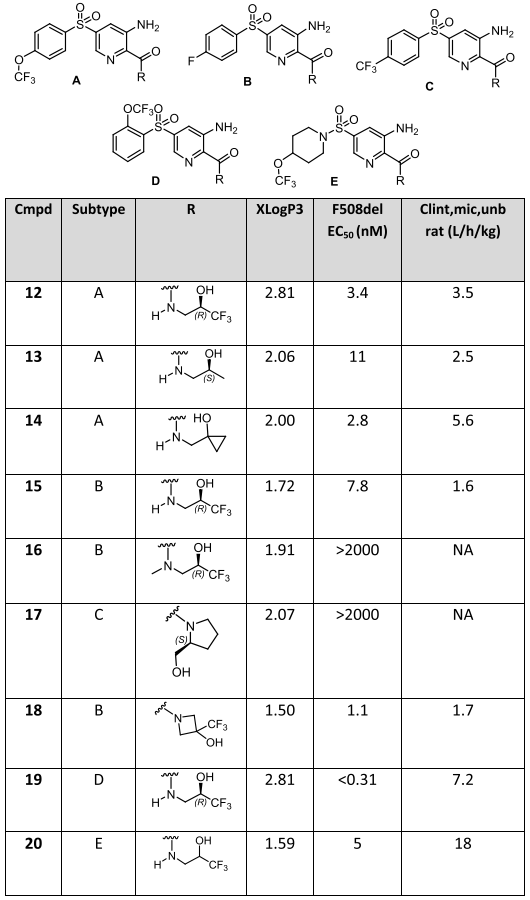
圖8 以化合物11為模版的結構優化
結合藥效和清除率,選擇化合物12、13、15、19、20進行大鼠PK評估(圖9)。數據表明,化合物12的半衰期>50h,這與總體低的清除率和較高的Vss有關,其非結合血漿清除率為5.2L/h/kg,說明該化合物具有良好的內在穩定性。化合物13和15在大鼠體內具有與化合物12相似的血漿清除率,但它們較低的Vss降低了靜脈給藥后的半衰期,分別為8.9h和>15h。清除率較高的化合物19和20分別給出了6.7h和1.3h的半衰期,結合次納摩爾級的藥效,化合物19被認為是比較有潛力的分子。
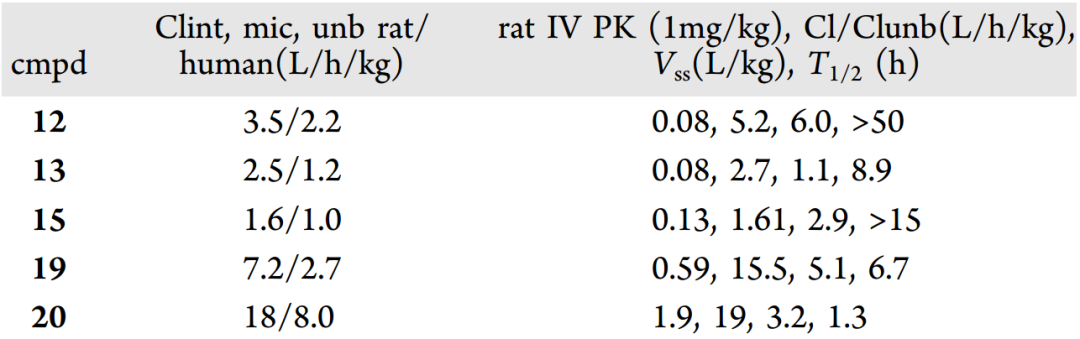
圖9 幾種化合物的大鼠PK數據
要實現成功的聯合治療,潛在的藥物-藥物相互作用(DDI)是一個必須考慮的因素,而與CYPP450酶的潛在相互作用通常被認為是最相關的相互作用,因此需要評估化合物對CYP的抑制和誘導作用。在噻吩甲酸酰胺類和3-氨基吡啶酰胺類這一系列化合物中,CYP抑制并不是普遍問題,通過PXR(孕烷X受體)報告發現,反而存在一些具有CYP誘導作用的化合物。基于上述化合物的大鼠PK數據,選擇化合物11、12、13、19,以Rifampicin為陽性對照,測試了它們對PXR的活化數據(圖10)。結果顯示,化合物13對PXR的活化相比對照組僅有12%,雖然化合物12擁有和化合物13相似的效果,但動力學水溶性較差。

圖10 幾種化合物對PXR的活化數據
基于化合物13的低CYP誘導潛力及其在藥效和大鼠PK方面整體的優異數據,進一步對該化合物進行了表征。在攜帶F508del突變患者的HBE細胞上測得該化合物的EC50為18nM,此外,該化合物在FaSSIF(空腹狀態下的人工胃液)介質中的熱力學水溶性為83μM,在MDCK實驗中展示出良好的滲透性,對CYP無明顯抑制作用,在MNT和AMES試驗中均為陰性,且未檢測到對hERG的抑制作用(圖11)。在狗和猴的PK試驗中,化合物13表現出較低的清除率,使得半衰期分別達到了15和16小時,而且口服給藥5mg/kg后,兩種動物的生物利用度均超過了90%(圖11)。


圖11 化合物13的綜合評估
根據以上實驗結果,將化合物13命名為GLPG2451并進行臨床前安全評估。
本文通過對增效劑3進行結構改造,將其中的噻吩酰胺骨架替換成吡啶酰胺,繼而優化篩選出新型增效劑GLPG2451。表征結果顯示,GLPG2451在攜帶F508del突變患者的HBE細胞上展現出優秀的生物活性,此外,該化合物在FaSSIF介質中的水溶性良好,對CYP無明顯抑制和誘導作用,MDCK細胞滲透性良好,無心臟毒性,而且骨架的替換避免了噻吩衍生物潛在的體外遺傳毒性。新型增效劑GLPG2451和增效劑GLPG1837雖然由同一初始分子衍生而來,但兩者的化學性質是不同的,最重要的是,相比Ivacaftor和GLPG1837需要每天2次給藥,GLPG2451只需要每天1次給藥。
StevenE. Van der Plas, Hans Kelgtermans, Oscar Mammoliti, Christel Menet,Giovanni Tricarico, Ann De Blieck, Caroline Joannesse, Tom De Munck,Dominique Lambin, Marlon Cowart, Sebastien Dropsit, Sebastien L. X.Martina, Maarten Gees, Anne-Sophie Wesse, Katja Conrath and MartinAndrews, Journalof Medicinal Chemistry(2021),64(1), 343-353.
注:文中圖片均來源于參考文獻
上一篇:手性二胺配體
下一篇:喜報!樂研成為中國化學試劑工業協會正式會員單位
 技術支持
技術支持



 400-821-0725
400-821-0725
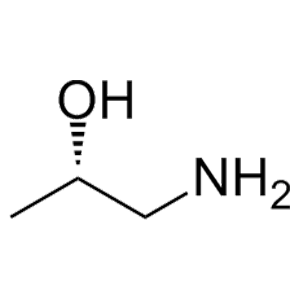
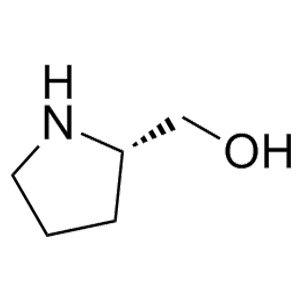
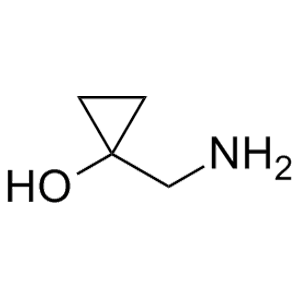
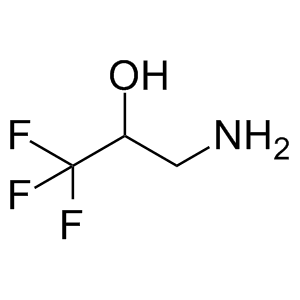

 滬ICP備17019645號
滬ICP備17019645號







